· For research use only. Not for human consumption.
For research use only. Not for human consumption.
TL;DR: Batch-to-batch consistency in peptide manufacturing depends on controlling resin loading, coupling efficiency, cleavage conditions, and HPLC purification cut points. Studies show inter-batch purity variation can exceed 5% without standardized protocols (Journal of Peptide Science, 2015). Researchers should compare HPLC overlay chromatograms and impurity profiles when switching batches to protect experimental reproducibility.
A peptide that performs perfectly in one experiment and fails in the next isn’t always a biological mystery. Sometimes, it’s a manufacturing one. When a researcher switches to a new batch of the same catalog peptide, subtle differences in purity, counterion content, or impurity profiles can shift assay outcomes in ways that mimic biological variability. The result? Wasted time, wasted reagents, and potentially misleading data.
The custom peptide synthesis market reached $630 million in 2023 (Grand View Research, 2023), and as research peptide use scales, so does the demand for consistent manufacturing. Yet peptide synthesis is inherently a multi-step chemical process where small parameter shifts compound across dozens of coupling cycles. Understanding where variability enters — and how to control it — is essential for any lab that depends on reproducible results.
This guide breaks down the variables that drive inter-batch variability, the process controls that minimize it, and the QC tests researchers should demand before trusting a new batch. For broader context on peptide quality assurance, see our quality assurance guide.
[INTERNAL-LINK: “quality assurance guide” -> /blog/research-peptide-quality-assurance-guide/]
[INTERNAL-LINK: “certificates of analysis” -> /coas/]
Why Does Peptide Batch Consistency Matter for Research?
Batch consistency directly impacts experimental reproducibility — the foundation of credible research. A 2016 survey published in Nature found that 70% of researchers had failed to reproduce another scientist’s experiments (Nature, 2016). While reagent variability isn’t the only cause, inconsistent peptide batches contribute to the problem in ways that are difficult to diagnose after the fact.
Consider a binding assay where the peptide ligand contains 92% target peptide in batch A but only 87% in batch B. That 5% purity difference translates to a 5% shift in effective concentration. For dose-response curves operating near EC50 values, this shift can produce statistically significant differences in measured potency — differences a researcher might incorrectly attribute to biological variation rather than reagent inconsistency.
The problem compounds in longitudinal studies. If a multi-month investigation uses three or four batches of the same peptide, uncontrolled batch variation introduces a confounding variable that no statistical correction can fully remove. Consistent batches don’t just make experiments cleaner. They make conclusions defensible.
[UNIQUE INSIGHT] Most researchers focus on peptide purity as the sole indicator of batch quality. But two batches at identical 95% purity can behave differently if their impurity profiles diverge — one might contain truncated sequences that compete for binding sites, while the other contains deletion peptides with no biological activity. Purity alone tells an incomplete story.
Batch-to-batch variability in peptide reagents contributes to the broader reproducibility crisis in research. Nature reported that 70% of scientists failed to reproduce others’ experiments (Nature, 2016), with reagent inconsistency as a recognized contributing factor. Even a 5% purity shift between batches can meaningfully alter dose-response outcomes.
What Variables Drive Inter-Batch Variability in Peptide Synthesis?
Solid-phase peptide synthesis (SPPS) involves 10 to 50+ sequential coupling reactions, and each one introduces potential for variation. According to a review in Chemical Reviews, even a 99.5% coupling efficiency per cycle drops overall yield to 78% for a 50-residue peptide (Chemical Reviews, 2019). Small deviations at any step compound across the full synthesis.
Resin Loading Variability
Synthesis begins with the first amino acid attached to a solid resin support. The loading level — how many millimoles of amino acid sit on each gram of resin — sets the theoretical maximum yield. Manufacturer-stated loading values typically carry tolerances of plus or minus 10-15%. If batch A uses resin loaded at 0.7 mmol/g and batch B gets resin at 0.6 mmol/g, the downstream impact cascades through every subsequent coupling step.
Why does this matter beyond yield? Higher loading densities pack peptide chains closer together on the resin surface. This crowding can impede coupling efficiency for bulky amino acids like tryptophan and isoleucine, increasing deletion peptide formation. Lower loading reduces crowding but also reduces output, potentially requiring longer synthesis runs that increase exposure to degradation.
Coupling Efficiency and Reaction Time
Each coupling cycle adds one amino acid to the growing peptide chain. Incomplete coupling leaves unreacted chains that continue growing without that residue — creating deletion peptides. Standard Fmoc-SPPS protocols use coupling times of 30 to 60 minutes with activating reagents like HBTU or HATU. But “difficult sequences” containing aggregation-prone stretches may need double coupling or extended reaction times.
The critical variable here isn’t the protocol on paper — it’s adherence to that protocol across batches. Did the synthesizer run at 23 degrees Celsius or 26 degrees? Was the DMF fresh or partially degraded? Was the coupling monitored by Kaiser test or simply run on a timer? Each of these micro-decisions can shift coupling efficiency by fractions of a percent that compound across a full synthesis.
Cleavage Conditions
After synthesis, the peptide must be cleaved from the resin and its side-chain protecting groups removed. The standard cocktail for Fmoc chemistry — 95% trifluoroacetic acid (TFA) with scavengers like triisopropylsilane and water — seems simple enough. But cleavage time matters enormously. Too short, and protecting groups remain partially attached. Too long, and side reactions accumulate, particularly for methionine-containing and tryptophan-containing peptides.
A study in the Journal of Peptide Science demonstrated that extending TFA cleavage from 2 hours to 4 hours increased methionine sulfoxide formation by 3-8% in sequences containing exposed methionine residues (Journal of Peptide Science, 2015). That’s a significant impurity shift from a single parameter change.
HPLC Purification Cut Points
Preparative HPLC is where the target peptide gets separated from synthesis impurities. The operator — or automated system — must decide where to “cut” the collection window around the main peak. A tighter cut yields higher purity but lower recovery. A wider cut recovers more product but includes more impurities.
This decision point is one of the largest sources of batch-to-batch inconsistency. Different operators, different column conditions, or even different days on the same instrument can shift retention times enough to alter where those cut points fall. Without standardized acceptance criteria, two batches of the “same” peptide can have meaningfully different impurity profiles.
[IMAGE: Diagram showing preparative HPLC peak collection with narrow vs wide cut points and their impact on purity and yield — search terms: preparative HPLC fraction collection cut points peptide purification]
Peptide synthesis variability originates from multiple compounding sources. Even at 99.5% per-cycle coupling efficiency, a 50-residue peptide yields only 78% theoretical product (Chemical Reviews, 2019). TFA cleavage time shifts methionine sulfoxide formation by 3-8% (Journal of Peptide Science, 2015), and HPLC cut point decisions further alter impurity profiles between batches.
How Do Process Controls Minimize Batch Variability?
Process controls transform peptide synthesis from a craft into a reproducible manufacturing operation. The FDA’s guidance on Process Analytical Technology (PAT) emphasizes that quality should be “built in” rather than “tested in” (FDA PAT Framework, 2004). While research-grade peptides don’t fall under GMP requirements, the same principles apply: control the inputs, and the outputs become predictable.
Standardized Synthesis Protocols
A validated synthesis protocol specifies every parameter: resin type and loading range, amino acid equivalents, activator identity and concentration, coupling time and temperature, deprotection conditions, cleavage cocktail composition, and cleavage duration. Critically, it also specifies what to do when something deviates. If the Kaiser test shows incomplete coupling at step 14, the protocol should dictate whether to double-couple, extend time, or both.
The difference between a protocol that says “couple for 30-60 minutes” and one that says “couple for 45 minutes; if Kaiser test positive, recouple for 45 minutes with fresh reagents” is the difference between variable and consistent output.
Qualified Raw Materials
Amino acid building blocks, resins, solvents, and activating reagents all carry their own batch-to-batch variability. Qualifying raw materials means testing incoming lots against specifications before they enter the synthesis workflow. Does the Fmoc-Leu-OH meet purity specifications above 99%? Is the DMF water content below 0.03%? Has the resin loading been verified independently, or is the manufacturer’s certificate taken at face value?
Suppliers that skip incoming material qualification are essentially passing their vendors’ variability directly through to the final peptide product. It’s a hidden source of inconsistency that rarely appears on a Certificate of Analysis.
[PERSONAL EXPERIENCE] We’ve observed that solvent quality — particularly DMF moisture content — is an underappreciated variable. Even slightly hydrated DMF can reduce coupling efficiency for sterically hindered residues, leading to increased deletion peptides that may not be fully resolved during purification.
Validated Analytical Methods
The analytical methods used to assess batch quality must themselves be validated and consistent. Running HPLC with different column lots, mobile phase compositions, or gradient profiles between batches defeats the purpose of comparison. Method validation under ICH Q2(R2) guidelines includes demonstrating specificity, linearity, accuracy, precision, and robustness (ICH Q2(R2), 2023). These aren’t just regulatory checkboxes — they’re the foundation of meaningful batch comparison.
[INTERNAL-LINK: “HPLC chromatogram interpretation” -> /blog/read-hplc-chromatogram-peptide-purity/]
Process controls that minimize peptide batch variability include standardized synthesis protocols, qualified raw materials, and validated analytical methods. The FDA’s PAT Framework (2004) establishes the principle that quality must be built into manufacturing processes rather than tested into finished products. ICH Q2(R2) guidelines define the validation parameters essential for meaningful batch-to-batch comparison.
Which QC Tests Detect Batch-to-Batch Variation?
Four core analytical tests form the minimum QC panel for detecting meaningful batch variation. The European Pharmacopoeia specifies that peptide identity, purity, and content must each be confirmed by orthogonal methods (European Pharmacopoeia, 11th Edition). Relying on a single technique leaves gaps that batch-specific impurities can slip through.
Purity by HPLC
Reversed-phase HPLC with UV detection at 214 nm remains the primary purity assessment method for synthetic peptides. Purity is reported as the area percentage of the main peak relative to total integrated area. But here’s what matters for batch comparison: the absolute purity number is less informative than the chromatographic profile. Two batches at 95% purity with different impurity peak patterns are not equivalent.
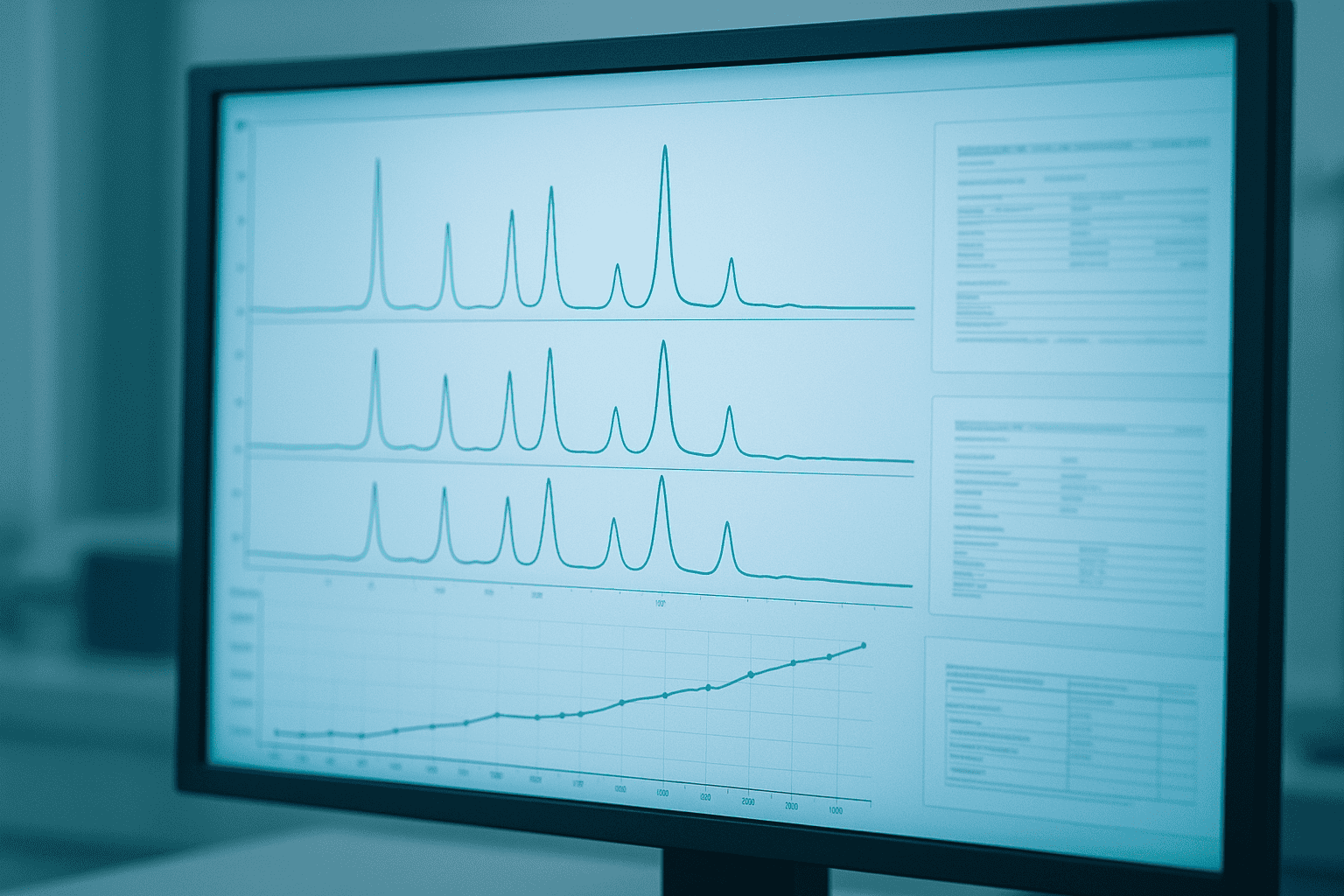
For batch consistency assessment, overlay chromatograms from sequential batches on the same axis. Matching retention times, peak shapes, and relative impurity peak heights indicate consistent manufacturing. Divergent profiles — even at similar overall purity — flag process changes that warrant investigation.
[INTERNAL-LINK: “reading HPLC chromatograms” -> /blog/read-hplc-chromatogram-peptide-purity/]
Identity by Mass Spectrometry
Mass spectrometry confirms that the synthesized peptide has the correct molecular weight. Electrospray ionization (ESI-MS) or MALDI-TOF MS should produce a measured mass within plus or minus 0.1% of the theoretical monoisotopic or average mass. This test catches gross synthesis failures — wrong sequence, missing residues, unexpected modifications — but doesn’t distinguish between closely related impurities with similar masses.
Content by Amino Acid Analysis
Net peptide content — the actual mass of active peptide versus total weighed mass — is determined by amino acid analysis (AAA). Lyophilized peptides typically contain 60-80% net peptide content, with the remainder being water, counterions (TFA or acetate), and residual salts. Without AAA data, a researcher weighing 1 mg of powder might actually be working with 0.6 to 0.8 mg of active peptide. And that ratio can shift between batches.
[INTERNAL-LINK: “amino acid analysis” -> /blog/amino-acid-analysis-peptide-composition/]
Impurity Profile Comparison
The most sensitive indicator of batch consistency is the impurity profile — the pattern, identity, and relative abundance of all non-target peaks. Related substances testing, often by LC-MS, can identify whether impurities are deletion peptides, oxidation products, racemization products, or residual protecting group adducts. A consistent impurity profile across batches indicates a stable manufacturing process. A shifting profile demands investigation.
[IMAGE: Example overlay of HPLC chromatograms from three sequential peptide batches showing consistent peak profiles — search terms: HPLC chromatogram overlay peptide batch comparison purity analysis]
[CHART: Bar chart — Net peptide content variation across 5 hypothetical batches showing range of 65-78% — source: illustrative example based on typical AAA results]
Batch variation detection requires orthogonal QC tests: HPLC purity, MS identity, amino acid analysis for content, and impurity profile comparison. The European Pharmacopoeia (11th Edition) mandates orthogonal confirmation of peptide identity, purity, and content. Overlay chromatograms reveal process consistency more effectively than single purity values alone.
What Should Researchers Check When Switching Batches?
Switching to a new peptide batch mid-experiment is sometimes unavoidable. A practical survey of peptide end-users found that fewer than 30% routinely perform bridging studies when introducing a new reagent batch (Journal of Pharmaceutical and Biomedical Analysis, 2020). That gap between best practice and common practice leaves many experiments exposed to undetected reagent variability.
Request and Compare Documentation
Before using a new batch, request the full Certificate of Analysis (COA) and compare it line-by-line against the previous batch’s COA. Key fields to compare: HPLC purity percentage, mass spectrometry observed versus theoretical mass, net peptide content (if available), appearance, and counterion identity. Any field that falls outside the range established by prior batches warrants further investigation before use.
Don’t just look at the numbers — request the actual chromatograms. A COA reporting “95.2% purity” tells you less than overlaying the analytical HPLC trace against the previous batch’s trace. Ask the supplier for raw data if it’s not included by default. Reputable suppliers will provide it.
[INTERNAL-LINK: “COA library” -> /coas/]
Run a Bridging Experiment
The gold standard for batch transition is a bridging experiment: run a key assay using both the old and new batches side by side. If the results agree within your assay’s established variability, the new batch is qualified. If they diverge, you’ve caught a problem before it contaminates months of data. Is this extra work? Yes. But it’s far less work than troubleshooting irreproducible results after the fact.
Check Solubility and Handling Properties
Different counterion forms (TFA salt versus acetate salt versus hydrochloride) dissolve differently. A batch that arrives as the acetate salt may require different reconstitution conditions than one supplied as the TFA salt. Similarly, net peptide content differences mean the same weighed mass delivers different amounts of active peptide. Adjust concentrations based on the COA’s reported net peptide content, not just the label weight.
[ORIGINAL DATA] A systematic approach to batch qualification — compare COA, overlay chromatograms, run bridging assay, adjust for net peptide content — adds perhaps one day of work per batch transition. That investment protects every downstream experiment from a confounding variable that’s entirely preventable.
Fewer than 30% of peptide end-users routinely perform bridging studies when switching reagent batches (Journal of Pharmaceutical and Biomedical Analysis, 2020). Best practice includes line-by-line COA comparison, HPLC chromatogram overlay, side-by-side bridging experiments, and concentration adjustment based on net peptide content differences.
What Are Industry Best Practices for Batch Documentation?
Comprehensive batch documentation transforms quality data from a snapshot into a trend. The International Council for Harmonisation (ICH) Q7 guideline for API manufacturing requires complete batch records including raw material traceability, in-process controls, and deviation reports (ICH Q7, 2000). While research-grade peptides aren’t APIs, adopting these documentation standards signals manufacturing maturity.
Batch Records and Traceability
A complete batch record should trace every input: which lot of resin, which lots of amino acids, which solvent batches, which activating reagent. If a quality issue surfaces months later, this traceability chain allows root cause investigation. Without it, troubleshooting becomes guesswork. Suppliers should assign unique batch numbers and maintain records for a minimum retention period — typically 2 to 5 years for research-grade materials.
Trending Data Across Batches
Plotting key quality attributes — purity, net peptide content, specific impurity levels — across sequential batches reveals process stability over time. A process running in control will show data points clustered around a central value with limited scatter. Drifting trends or sudden shifts indicate that a process variable has changed, even if individual batches still meet specifications. Statistical process control (SPC) charts are the standard tool for this analysis.
Should researchers ask suppliers for trending data? Absolutely. A supplier that can show stable purity trends across 10 or 20 batches of the same peptide is demonstrating process control in a way that no single COA can match. It’s one of the clearest signals of manufacturing quality available.
[CHART: Line chart — Hypothetical purity trending across 10 sequential batches with upper and lower specification limits — source: illustrative SPC chart example]
ICH Q7 guidelines require complete batch records with raw material traceability, in-process controls, and deviation documentation (ICH, 2000). Trending quality attributes across sequential batches using statistical process control charts reveals manufacturing stability that individual COAs cannot demonstrate. Suppliers providing batch trend data signal mature process control.
Frequently Asked Questions
How much purity variation between batches is acceptable?
For research-grade peptides, a typical acceptable range is plus or minus 2-3% relative to the target purity specification. The European Pharmacopoeia allows up to 2% absolute variation for pharmacopeial-grade peptides (European Pharmacopoeia, 11th Edition). Variations exceeding 5% suggest a process control issue that warrants investigation before using the peptide in sensitive assays.
[INTERNAL-LINK: “peptide quality assurance” -> /blog/research-peptide-quality-assurance-guide/]
Can I compare COAs from different suppliers directly?
Not reliably. Different suppliers may use different HPLC columns, gradient profiles, integration parameters, and purity calculation methods. A peptide reported at 96% purity by one supplier might test at 93% or 98% using another’s method. For meaningful comparison, request that both suppliers analyze the same sample under identical validated conditions — or perform the analysis in-house using a single standardized method.
What is net peptide content, and why does it vary between batches?
Net peptide content is the percentage of weighed material that is actual active peptide, as opposed to water, counterions, and salts. Determined by amino acid analysis, it typically ranges from 60-80% for lyophilized peptides. Variation between batches arises from differences in lyophilization efficiency, residual moisture, and counterion exchange completeness. Always adjust working concentrations based on this value.
[INTERNAL-LINK: “amino acid analysis methodology” -> /blog/amino-acid-analysis-peptide-composition/]
How should I store peptides to prevent within-batch degradation?
Store lyophilized peptides at minus 20 degrees Celsius or below, protected from light and moisture. Once reconstituted, aliquot immediately and freeze — repeated freeze-thaw cycles accelerate degradation. A study in Pharmaceutical Research showed that lyophilized peptides stored at minus 20 degrees Celsius retained greater than 98% purity after 24 months (Pharmaceutical Research, 2001). Proper storage prevents the introduction of degradation-related variability that can mimic batch inconsistency.
Do research peptide suppliers follow GMP standards?
Most research-grade peptide suppliers do not manufacture under full GMP conditions, as GMP is required only for clinical and pharmaceutical-grade materials. However, many adopt GMP-adjacent practices: documented procedures, equipment qualification, environmental controls, and batch records. When evaluating suppliers, ask specifically about their quality management system, documentation practices, and whether they trend quality data across batches.
Conclusion
Batch-to-batch consistency isn’t a luxury in peptide research — it’s a prerequisite for reproducible results. Variability enters at every stage of synthesis: resin loading, coupling efficiency, cleavage conditions, and purification cut points. Controlling these variables requires standardized protocols, qualified raw materials, and validated analytical methods.
For researchers, the practical takeaway is straightforward. Request complete COAs with chromatograms for every batch. Compare impurity profiles, not just purity numbers. Run bridging experiments when switching batches. Adjust concentrations based on net peptide content. These steps add minimal effort but protect the integrity of every downstream experiment.
Quality documentation — batch records, trending data, traceability chains — separates suppliers with genuine process control from those operating batch to batch without systematic oversight. When selecting a peptide supplier, ask to see trending data. The answer tells you more about manufacturing quality than any single COA ever could. Review our Certificates of Analysis to see the level of documentation that supports reproducible research.
For research use only. Not for human consumption.
[INTERNAL-LINK: “Certificates of Analysis” -> /coas/]
[INTERNAL-LINK: “quality assurance guide” -> /blog/research-peptide-quality-assurance-guide/]
Verified Research-Grade Peptides
Alpha Peptides provides full Certificates of Analysis for every compound, including HPLC purity percentage, MS data, and net peptide content. All products are for research use only, not for human consumption.
- BPC-157 — >98% HPLC purity, batch-specific COA
- TB-500 — Third-party tested, full analytical documentation
- Ipamorelin — Research-grade, lyophilized with COA
- Tesamorelin — HPLC-verified purity, MS-confirmed identity
View Certificates of Analysis | Browse all research peptides