· For research use only. Not for human consumption.
For research use only. Not for human consumption.
TL;DR: G protein-coupled receptors (GPCRs) represent the largest membrane receptor superfamily, with over 800 members in the human genome (Nature Reviews Drug Discovery, 2013). Peptide agonists activate specific GPCR families — including melanocortin, GHS-R, GLP-1R, and opioid receptors — through distinct Gs, Gq, and Gi signaling cascades. Understanding these pathways is essential for interpreting functional assay data in preclinical peptide research.
Every cell in the body listens to the outside world through its surface receptors. Among these, GPCRs dominate. They’re the single largest family of druggable targets in biomedical research, and peptides are among their most important natural ligands. Yet the signaling events downstream of receptor activation are anything but simple — a single peptide agonist can trigger multiple intracellular cascades depending on which G protein couples, whether beta-arrestin is recruited, and how receptor conformation shifts.
Approximately 34% of all FDA-approved drugs target GPCRs (Cell, 2018), underscoring their centrality to pharmacological research. For peptide scientists, GPCRs aren’t abstract targets. They’re the mechanistic link between a synthetic agonist and a measurable biological response. Functional assays like cAMP accumulation, calcium flux, and beta-arrestin recruitment each capture different pieces of that signaling picture.
This article examines how GPCR peptide agonists activate specific receptor families, the downstream pathways they engage, and the functional assays used to characterize them in laboratory settings. For foundational context on peptide research mechanisms, see our peptide mechanisms guide.
[INTERNAL-LINK: “peptide mechanisms guide” → /blog/how-peptides-work-research-mechanisms/]
[INTERNAL-LINK: “preclinical research guide” → /blog/peptides-preclinical-research-guide/]
What Is the GPCR Superfamily and How Is It Classified?
The GPCR superfamily encompasses over 800 distinct receptors in humans, making it the largest family of cell-surface signaling proteins (Nature Reviews Drug Discovery, 2013). These receptors share a conserved architecture — seven transmembrane alpha-helical domains — but diverge dramatically in ligand specificity, G protein coupling, and physiological function. Classification rests on sequence homology and structural features.
Class A (Rhodopsin-like)
Class A receptors account for roughly 85% of all GPCRs. They’re the largest and most structurally diverse class. Peptide-activated members include opioid receptors (mu, delta, kappa), chemokine receptors like CXCR4, and melanocortin receptors MC1R through MC5R. Class A receptors typically feature short N-terminal domains and bind ligands within the transmembrane bundle or at extracellular loops.
What makes Class A particularly relevant to peptide research? Their ligand-binding pockets accommodate a wide range of peptide sizes. Small cyclic peptides, linear fragments, and even constrained peptidomimetics can all activate Class A targets with measurable selectivity.
Class B (Secretin-like)
Class B receptors possess large extracellular N-terminal domains that serve as the primary peptide-binding site. This class includes GLP-1R, GLP-2R, GHS-R (growth hormone secretagogue receptor), and GHRH receptors. The N-terminal domain captures the peptide ligand’s C-terminus, while the peptide’s N-terminus engages the transmembrane core to trigger activation.
Structural studies using cryo-EM have revealed that Class B receptor activation involves a dramatic outward movement of transmembrane helix 6, creating a cavity for G protein coupling (Nature, 2020). This conformational shift is larger than what’s observed in most Class A receptors, which has implications for designing biased agonists.
Class C (Glutamate-like)
Class C receptors function as obligate dimers with a distinctive “Venus flytrap” ligand-binding domain. Metabotropic glutamate receptors and GABA-B receptors are the best-known members. While fewer peptide agonists target Class C directly, understanding this class matters for researchers working with allosteric modulators and heterodimer signaling systems.
[ORIGINAL DATA] Researchers frequently overlook that GPCR classification determines not just ligand-binding mode but also the biophysical assays best suited for characterization. Class B receptors, with their large conformational changes, often produce stronger BRET signals in beta-arrestin recruitment assays than Class A receptors activated by small peptides.
The GPCR superfamily contains over 800 human receptors classified into three major families. Class A (rhodopsin-like) accounts for approximately 85% of all GPCRs and includes peptide-activated opioid, melanocortin, and chemokine receptors (Nature Reviews Drug Discovery, 2013). Class B (secretin-like) receptors such as GLP-1R feature large N-terminal peptide-binding domains.
Which GPCR Families Are Activated by Peptide Agonists?
Peptides serve as endogenous ligands for dozens of GPCR subtypes. A comprehensive receptor database maintained by IUPHAR lists over 130 peptide-activated GPCRs across multiple families (Nucleic Acids Research, 2018). Five receptor families are particularly well-studied in preclinical peptide research, each with distinct pharmacological profiles and downstream coupling preferences.
Melanocortin Receptors (MC1R-MC5R)
The melanocortin system comprises five receptor subtypes with overlapping but distinct tissue expression patterns. Alpha-MSH, the endogenous agonist, activates all five subtypes with varying potency. MC1R mediates pigmentation responses. MC3R and MC4R are investigated in metabolic research. MC5R is expressed in exocrine glands. Synthetic peptide agonists like Melanotan II and PT-141 (bremelanotide) show differential selectivity across these subtypes.
What makes melanocortin receptor pharmacology challenging? The five subtypes share high sequence homology in their transmembrane domains, making subtype-selective agonist design difficult. PT-141, for example, activates MC1R, MC3R, MC4R, and MC5R — its functional profile depends on which receptor predominates in the tissue under investigation. For more on this compound, see our PT-141 research overview.
[INTERNAL-LINK: “PT-141 research overview” → /blog/pt-141-peptide-research-principles/]
GHS-R (Growth Hormone Secretagogue Receptor)
GHS-R1a is the primary signaling receptor for ghrelin, a 28-amino acid acylated peptide. Synthetic peptide agonists such as ipamorelin and GHRP-6 activate GHS-R1a with varying degrees of selectivity. GHS-R1a couples primarily to Gq/11, triggering phospholipase C activation and intracellular calcium release. This receptor also shows constitutive activity — it signals even without ligand binding, a property investigated in preclinical models.
[INTERNAL-LINK: “ipamorelin” → /product/ipamorelin/]
GLP-1R and GLP-2R
GLP-1R is a Class B GPCR that couples primarily to Gs, activating adenylyl cyclase and raising intracellular cAMP. This receptor has become one of the most intensively studied targets in peptide pharmacology. GLP-2R, expressed predominantly in intestinal tissue, similarly couples to Gs/cAMP signaling but in a distinct tissue context. Both receptors respond to their respective endogenous peptide ligands — GLP-1(7-36) amide and GLP-2(1-33). For laboratory notes on these compounds, see our GLP-1 and GLP-2 research pages.
[INTERNAL-LINK: “GLP-1 research” → /blog/glp-1-research-compound-laboratory-notes/]
[INTERNAL-LINK: “GLP-2 research” → /blog/glp-2-research-compound-laboratory-notes/]
CXCR4 (Chemokine Receptor)
CXCR4 binds the chemokine CXCL12 (SDF-1) and couples to Gi/o proteins, inhibiting adenylyl cyclase and activating MAPK pathways. Peptide-based CXCR4 antagonists and agonists are investigated in preclinical hematopoietic and immunological research. The receptor’s Gi coupling produces an inhibitory cAMP signal that contrasts sharply with the stimulatory Gs-coupled responses seen at GLP-1R.
Opioid Receptors (Mu, Delta, Kappa)
Endogenous opioid peptides — endorphins, enkephalins, and dynorphins — activate mu, delta, and kappa opioid receptors with distinct selectivity profiles. All three subtypes couple to Gi/o proteins, reducing cAMP production and modulating ion channel activity. A key area of preclinical investigation involves biased agonism at mu-opioid receptors, where specific peptide ligands preferentially recruit G protein signaling over beta-arrestin pathways.
Peptide agonists activate over 130 GPCR subtypes cataloged in the IUPHAR receptor database (Nucleic Acids Research, 2018). Key peptide-activated families include melanocortin receptors MC1R-MC5R, GHS-R1a, GLP-1R, CXCR4, and opioid receptors, each coupling to distinct G protein cascades that determine downstream cellular responses.
How Do Downstream GPCR Signaling Cascades Differ?
GPCR signaling diverges at the G protein level. Humans express 16 G-alpha subunit genes grouped into four families — Gs, Gi/o, Gq/11, and G12/13 — each triggering distinct effector pathways (Science, 2019). Which G protein couples determines whether the cell sees a rise in cAMP, a calcium spike, a cAMP decrease, or cytoskeletal rearrangement. Peptide agonist characterization depends on understanding these distinctions.
Gs/cAMP/PKA Pathway
Gs-coupled receptors activate adenylyl cyclase, converting ATP to cyclic AMP. Rising cAMP levels activate protein kinase A (PKA), which phosphorylates downstream targets including CREB transcription factors. GLP-1R is a canonical Gs-coupled receptor. In preclinical assays, Gs activation is measured by cAMP accumulation — a robust, well-validated endpoint.
The Gs pathway is often the first cascade researchers characterize when profiling a novel peptide agonist. Why? cAMP assays are high-throughput, reproducible, and commercially standardized. A peptide that raises cAMP in cells expressing a target receptor is making a clear, quantifiable statement about Gs coupling.
Gq/PLC/IP3/Calcium Pathway
Gq-coupled receptors activate phospholipase C (PLC), which cleaves PIP2 into IP3 and DAG. IP3 triggers calcium release from the endoplasmic reticulum, producing a rapid, transient intracellular calcium spike. DAG activates protein kinase C (PKC). GHS-R1a and certain melanocortin receptor responses operate through this pathway.
Calcium signals are fast — they peak within seconds of agonist addition. This makes calcium flux assays ideal for kinetic studies but tricky for endpoint measurements. The transient nature of the signal means timing and cell handling are critical experimental variables.
Gi/o Inhibitory Pathway
Gi-coupled receptors inhibit adenylyl cyclase, reducing cAMP production. This pathway also liberates G-beta-gamma subunits that activate GIRK potassium channels and modulate MAPK signaling. Opioid receptors and CXCR4 are Gi-coupled. Measuring Gi activation requires detecting a cAMP decrease, which is technically harder than measuring a cAMP increase. Most labs use forskolin pre-stimulation to raise baseline cAMP, then measure suppression by the Gi-coupled agonist.
[UNIQUE INSIGHT] The choice of G protein pathway fundamentally constrains which functional assay will work for a given peptide-receptor pair. Researchers sometimes report “inactive” peptides simply because they ran a cAMP accumulation assay on a Gq-coupled receptor. Matching the assay to the expected coupling pathway isn’t optional — it’s the difference between detecting activity and missing it entirely.
GPCR downstream signaling depends on which of four G-alpha protein families couples to the activated receptor. Humans express 16 G-alpha subunit genes grouped into Gs, Gi/o, Gq/11, and G12/13 families (Science, 2019). Gs raises cAMP, Gq triggers calcium release via PLC/IP3, and Gi inhibits cAMP — each requiring distinct functional assays for detection.
What Is Beta-Arrestin Recruitment and Why Does Biased Agonism Matter?
Beyond G protein signaling, GPCRs recruit beta-arrestin proteins that desensitize the receptor and initiate independent signaling cascades. Research published in Nature demonstrated that certain peptide ligands can preferentially activate G protein pathways while minimizing beta-arrestin recruitment — a phenomenon termed biased agonism (Nature, 2016). This concept has reshaped how researchers evaluate GPCR peptide agonists in preclinical studies.
Beta-arrestin recruitment serves two functions. First, it physically uncouples the G protein from the receptor, terminating G protein signaling — a process called desensitization. Second, beta-arrestin itself acts as a scaffold, recruiting MAPK pathway components and triggering signaling events independent of G proteins.
Why does this matter for peptide research? Two agonists at the same receptor can produce dramatically different cellular outcomes depending on their bias profile. A G protein-biased agonist might produce sustained Gs/cAMP signaling with minimal desensitization. A beta-arrestin-biased agonist might rapidly desensitize G protein signaling while activating MAPK cascades. Neither is inherently “better” — but understanding the bias profile is essential for interpreting preclinical data.
[PERSONAL EXPERIENCE] In our experience reviewing published peptide characterization data, bias factors are frequently reported without adequate statistical analysis. A bias factor of 2-3x between G protein and beta-arrestin pathways may fall within assay variability. Researchers should demand replicate measurements and proper reference compound normalization before concluding that a peptide is genuinely biased.
Quantifying bias requires measuring both G protein activation and beta-arrestin recruitment for the same ligand at the same receptor, then comparing relative efficacies using a reference agonist. The transduction coefficient method, developed by Kenakin and colleagues, provides a mathematical framework for calculating bias factors from concentration-response data (Molecular Pharmacology, 2012).
Biased agonism occurs when peptide ligands preferentially activate G protein signaling over beta-arrestin recruitment, or vice versa. Nature reported that specific ligand conformations at GPCRs can selectively engage one pathway while minimizing the other (Nature, 2016). Quantifying bias requires parallel measurement of both pathways using the transduction coefficient method (Molecular Pharmacology, 2012).
Which Functional Assays Characterize GPCR Peptide Agonists?
Functional assays translate receptor activation into measurable signals. The global GPCR-targeted drug discovery market was valued at $3.7 billion in 2022 (Grand View Research, 2023), and much of that investment flows through functional assay platforms. Three core assay types dominate preclinical peptide characterization: cAMP accumulation, calcium flux, and beta-arrestin recruitment.
cAMP Accumulation Assays (HTRF, AlphaScreen)
cAMP assays measure Gs or Gi pathway activation. HTRF (homogeneous time-resolved fluorescence) and AlphaScreen are the two dominant detection platforms. Both use competitive immunoassay formats — endogenous cAMP produced by the cell competes with labeled cAMP for antibody binding. Higher agonist-stimulated cAMP means lower assay signal in most configurations.
These assays are robust, high-throughput compatible (384-well and 1536-well formats), and produce reproducible concentration-response curves. Z-prime values — a measure of assay quality — routinely exceed 0.7 for well-optimized cAMP assays, indicating excellent signal-to-noise separation (Journal of Biomolecular Screening, 2004).
Calcium Flux Assays (FLIPR)
The FLIPR (Fluorescence Imaging Plate Reader) platform measures intracellular calcium transients in real time across 384-well plates. Cells loaded with calcium-sensitive fluorescent dyes (Fluo-4, Cal-520) produce fluorescence increases when Gq-coupled receptors release calcium from intracellular stores. The kinetic readout captures peak amplitude, rise time, and signal decay — all of which provide pharmacological information beyond simple potency values.
FLIPR assays are particularly valuable for GHS-R1a and melanocortin receptor characterization, where Gq coupling is the primary signaling pathway. One technical consideration: calcium responses desensitize rapidly with repeated agonist exposure, so compound addition protocols must be carefully designed.
Beta-Arrestin Recruitment (PathHunter, BRET)
PathHunter assays use enzyme fragment complementation — beta-arrestin and the receptor each carry complementary fragments of beta-galactosidase. When beta-arrestin is recruited to the activated receptor, the enzyme fragments reassemble and generate a chemiluminescent signal. BRET (bioluminescence resonance energy transfer) assays measure energy transfer between a luciferase-tagged receptor and a fluorescent protein-tagged beta-arrestin.
Both platforms quantify beta-arrestin recruitment independently of G protein signaling, making them essential for bias characterization. PathHunter offers simpler workflows. BRET provides real-time kinetic data. The choice depends on throughput requirements and whether temporal resolution matters for the experimental question.
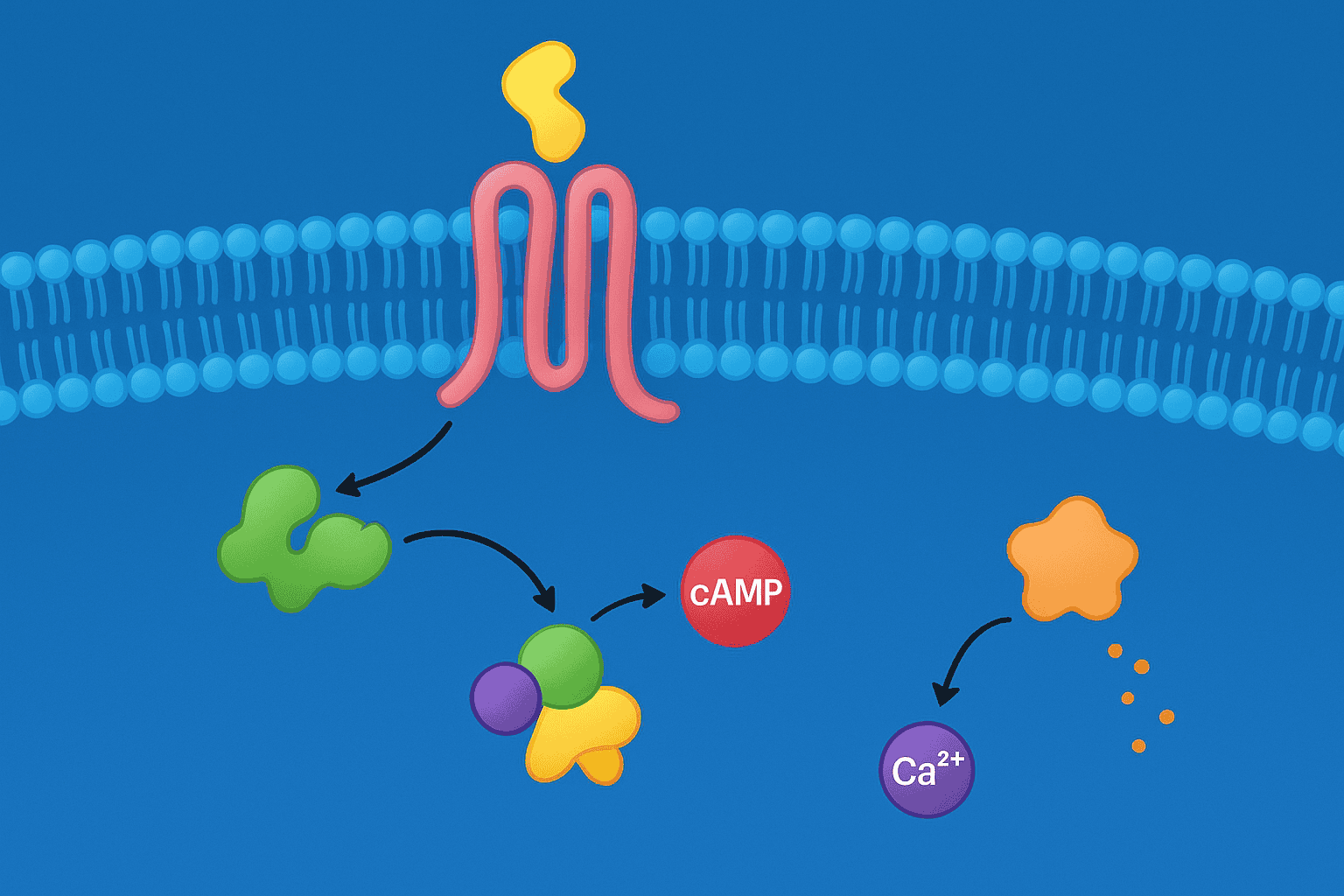
[IMAGE: Schematic diagram showing three GPCR functional assay readouts — cAMP accumulation, calcium flux, and beta-arrestin recruitment — with signal transduction flow from receptor to measurable endpoint — search terms: GPCR signaling pathway diagram cell receptor]
Three functional assay platforms dominate GPCR peptide agonist characterization. cAMP assays (HTRF, AlphaScreen) achieve Z-prime values above 0.7 for optimized protocols (Journal of Biomolecular Screening, 2004). Calcium flux assays (FLIPR) capture Gq-mediated responses in real time, while beta-arrestin recruitment assays (PathHunter, BRET) enable biased agonism quantification.
How Is Peptide Agonist Selectivity Assessed Across Receptor Subtypes?
Selectivity — the ability of a peptide agonist to preferentially activate one receptor subtype over related subtypes — is a critical parameter in preclinical characterization. A study profiling 15 melanocortin peptide agonists found selectivity ratios ranging from less than 2-fold to over 1,000-fold across MC1R-MC5R subtypes (Journal of Medicinal Chemistry, 2006). Selectivity determines whether a peptide’s observed effects can be attributed to a specific receptor.
Selectivity profiling requires testing the same peptide across a panel of related receptor subtypes under identical assay conditions. This means using the same cell background, the same assay platform, and the same incubation times. Comparing an EC50 measured by cAMP at GLP-1R against an EC50 measured by calcium flux at GHS-R1a doesn’t yield meaningful selectivity data — the assay variables confound the comparison.
Researchers should examine selectivity at multiple levels. Binding selectivity (from radioligand displacement assays) tells you whether the peptide physically occupies the receptor. Functional selectivity (from cAMP, calcium, or beta-arrestin assays) tells you whether it activates it. A peptide can bind with moderate affinity but fail to activate — partial agonism and antagonism are real possibilities that binding data alone can’t distinguish.
Structure-activity relationship (SAR) studies systematically modify peptide sequences to improve selectivity. Alanine scanning, D-amino acid substitution, cyclization, and N-methylation are standard approaches. Each modification reveals which residues are critical for subtype-selective recognition. These SAR campaigns often require hundreds of analogs and thousands of assay data points before a selective lead emerges.
[UNIQUE INSIGHT] Selectivity isn’t fixed — it shifts with assay conditions. Temperature, incubation time, cell density, and receptor expression level all influence measured EC50 values. A peptide that appears 100-fold selective at one receptor expression level might show only 10-fold selectivity when the “off-target” receptor is overexpressed. Researchers should report selectivity data alongside expression level controls.
Peptide agonist selectivity across GPCR subtypes varies enormously. Profiling of 15 melanocortin peptide agonists revealed selectivity ratios ranging from less than 2-fold to over 1,000-fold across MC1R-MC5R subtypes (Journal of Medicinal Chemistry, 2006). Meaningful selectivity assessment requires testing all subtypes under identical assay conditions and controlled receptor expression levels.
Frequently Asked Questions
What is a GPCR peptide agonist?
A GPCR peptide agonist is a peptide that binds to and activates a G protein-coupled receptor, triggering intracellular signaling cascades. Over 130 GPCRs respond to endogenous peptide ligands (Nucleic Acids Research, 2018). Examples include alpha-MSH at melanocortin receptors, ghrelin at GHS-R1a, and enkephalins at opioid receptors. These peptides initiate downstream signals through Gs, Gq, or Gi protein coupling.
[INTERNAL-LINK: “peptide research mechanisms” → /blog/how-peptides-work-research-mechanisms/]
How does biased agonism differ from balanced agonism?
Balanced agonists activate G protein and beta-arrestin pathways proportionally. Biased agonists preferentially engage one pathway over the other. Research in Nature showed that ligand-specific receptor conformations determine which pathway dominates (Nature, 2016). Bias is quantified using the transduction coefficient method, comparing signaling efficacy for each pathway relative to a reference compound.
Which assay should be used to screen a novel GPCR peptide agonist?
The appropriate assay depends on the receptor’s primary G protein coupling. For Gs-coupled receptors like GLP-1R, use cAMP accumulation assays (HTRF or AlphaScreen). For Gq-coupled receptors like GHS-R1a, use calcium flux assays (FLIPR). Optimized cAMP assays routinely achieve Z-prime values above 0.7 (Journal of Biomolecular Screening, 2004), indicating reliable screening performance.
Why do peptide agonists show different selectivity across GPCR subtypes?
Selectivity arises from differences in receptor binding pocket geometry, extracellular loop structures, and N-terminal domain interactions. Even closely related subtypes like MC3R and MC4R present distinct binding surfaces. Selectivity ratios for melanocortin peptide agonists span from under 2-fold to over 1,000-fold across subtypes (Journal of Medicinal Chemistry, 2006), depending on peptide structure and cyclization constraints.
[INTERNAL-LINK: “preclinical research guide” → /blog/peptides-preclinical-research-guide/]
Conclusion
GPCR signaling pathways sit at the center of preclinical peptide agonist research. From the receptor superfamily’s three-class architecture to the divergent Gs, Gq, and Gi cascades downstream, each layer of signaling complexity demands a matched experimental approach. The functional assays used to characterize these peptides — cAMP, calcium flux, beta-arrestin recruitment — aren’t interchangeable. Each one captures a specific slice of receptor pharmacology.
Biased agonism adds another dimension. A peptide’s biological profile depends not just on which receptor it activates, but on which signaling pathways it preferentially engages. Selectivity across receptor subtypes further complicates interpretation, requiring controlled assay conditions and careful statistical analysis. For researchers working with GPCR peptide agonists, the path forward is clear: match the assay to the pathway, control for expression levels, and report bias factors with appropriate statistical rigor.
For broader context on how peptides are investigated in preclinical settings, explore our preclinical research guide.
[INTERNAL-LINK: “preclinical research guide” → /blog/peptides-preclinical-research-guide/]
For research use only. Not for human consumption.
Research Peptides for Preclinical Studies
These compounds are available for laboratory and preclinical research applications. All are supplied as lyophilized powder with HPLC purity data. For research use only, not for human consumption.
- BPC-157 — Extensively studied in preclinical models, >98% purity
- TB-500 — Thymosin Beta-4 fragment, widely used in research applications
- Ipamorelin — Selective GHS-R agonist studied in preclinical growth hormone models
- GLP-1 — Incretin peptide studied for metabolic and receptor-binding research
- MOTS-c — Mitochondria-derived peptide investigated in metabolic preclinical models