· For research use only. Not for human consumption.
For research use only. Not for human consumption.
Why Is Peptide Lipidation a Critical Research Strategy?
Most unmodified peptides are cleared from circulation within minutes, severely limiting their utility in preclinical studies. Lipidation — the covalent attachment of fatty acid chains to peptide sequences — has emerged as one of the most effective strategies for extending peptide half-life. According to a 2020 review in Nature Reviews Drug Discovery, lipidated peptide analogs now represent over 30% of peptide-based compounds in advanced preclinical and clinical investigation stages.
This technique works by enabling reversible binding to serum albumin, which shields the peptide from enzymatic degradation and renal filtration. The result? Half-lives that extend from minutes to days in animal models. Researchers working with GLP-1 and GLP-2 receptor agonist analogs, as well as acylated amylin analogs like Cagrilintide, have examined lipidation extensively in laboratory settings.
[INTERNAL-LINK: peptide chemistry fundamentals → /blog/peptide-chemistry-guide/]
This article covers the chemistry behind fatty acid conjugation, the most common acyl chains used, conjugation strategies, analytical characterization methods, and how lipidation compares with PEGylation. All discussion reflects preclinical research data — not therapeutic claims.
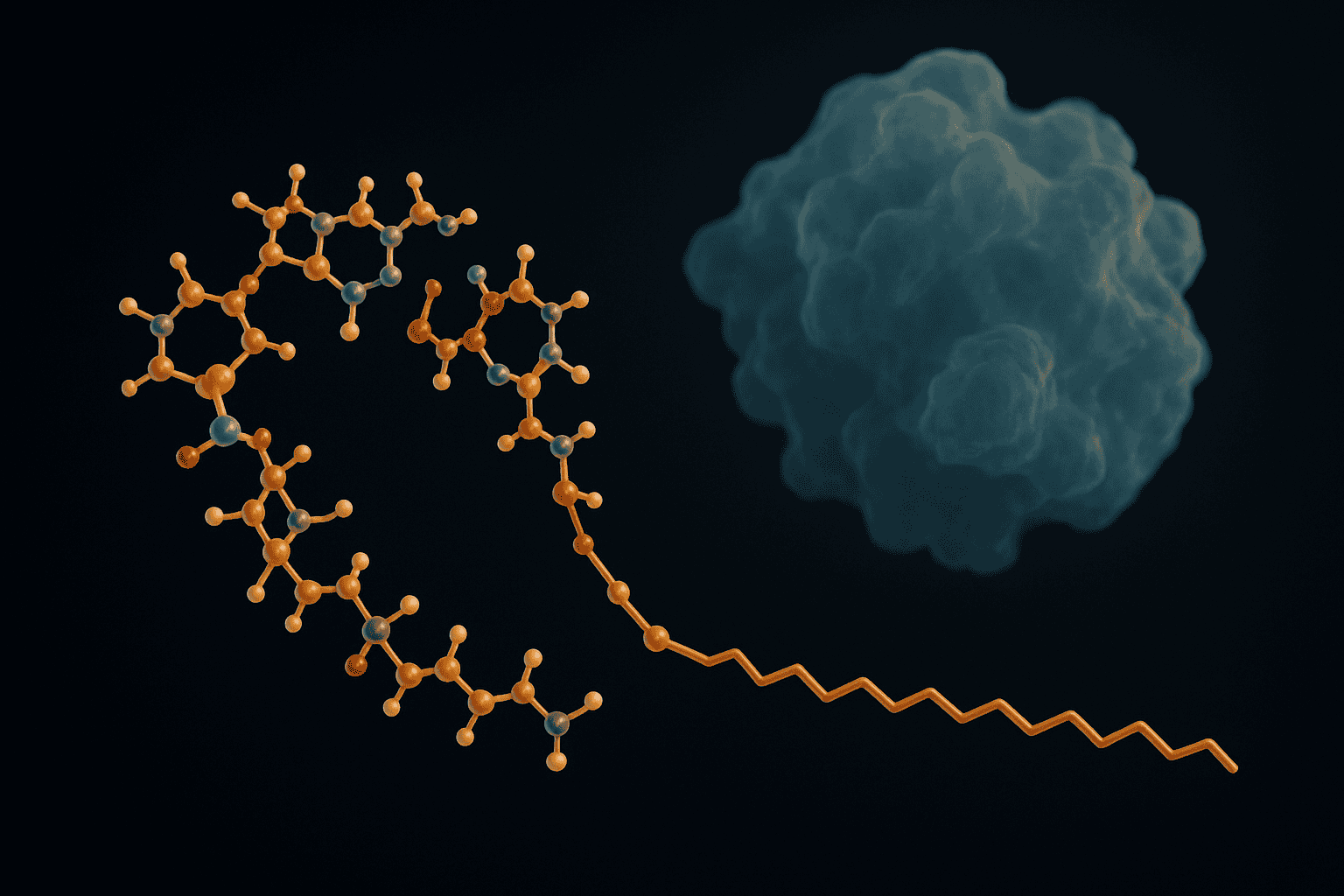
[IMAGE: Molecular structure diagram showing fatty acid chain conjugated to peptide backbone via lysine side chain — lipidated peptide molecular structure diagram acylation]
TL;DR: Peptide lipidation conjugates fatty acid chains (C16-C18) to peptide sequences, enabling reversible albumin binding that extends circulating half-life from minutes to days in preclinical models. Over 30% of peptide compounds in advanced investigation stages now use this approach (Nature Reviews Drug Discovery, 2020). Key methods include direct acylation and glutamic acid spacer strategies.
What Is Peptide Lipidation?
Peptide lipidation is the covalent attachment of a fatty acid moiety to a peptide chain, typically through an amide bond to a lysine side chain. A 2019 paper in Journal of Medicinal Chemistry described lipidation as the single most impactful post-synthetic modification for improving peptide pharmacokinetic profiles in preclinical models. The technique mimics natural lipid modifications found across biological systems.
Peptide lipidation — the covalent attachment of fatty acid chains to peptide sequences — is considered the single most impactful post-synthetic modification for improving peptide pharmacokinetic profiles in preclinical models. The technique enables reversible albumin binding, extending half-life from minutes to days (Journal of Medicinal Chemistry, 2019).
In biological systems, lipidation occurs naturally. Myristoylation, palmitoylation, and prenylation all serve to anchor proteins to cell membranes or modulate protein-protein interactions. Synthetic lipidation borrows this principle but applies it strategically — attaching specific fatty acid chains at defined positions to achieve albumin binding rather than membrane anchoring.
The distinction matters. Membrane-anchored peptides would remain localized at administration sites. Albumin-binding peptides circulate freely but are protected from degradation. Getting this balance right requires careful selection of the acyl chain length, attachment site, and spacer chemistry.
[INTERNAL-LINK: GLP-1 research compound background → /blog/glp-1-research-compound-laboratory-notes/]
Which Fatty Acid Chains Are Used in Lipidation Research?
Three acyl chains dominate the lipidation literature. According to a 2021 systematic review in European Journal of Pharmaceutics and Biopharmaceutics, palmitic acid (C16), stearic acid (C18), and octadecanedioic acid (C18 diacid) account for over 85% of lipidated peptide constructs examined in preclinical studies. Each chain length produces distinct pharmacokinetic behaviors.
Palmitic acid (C16), stearic acid (C18), and octadecanedioic acid (C18 diacid) account for over 85% of lipidated peptide constructs in preclinical studies. Longer acyl chains generally increase albumin binding affinity but can reduce aqueous solubility, requiring careful optimization (European Journal of Pharmaceutics and Biopharmaceutics, 2021).
Palmitic Acid (C16)
Palmitic acid, a 16-carbon saturated fatty acid, was among the earliest chains used for peptide lipidation. It provides moderate albumin binding affinity and good aqueous solubility relative to longer chains. Early acylated insulin analogs employed palmitic acid conjugation, establishing the foundation for modern lipidation strategies.
The C16 chain offers a practical balance. It’s long enough to bind albumin’s fatty acid binding sites but short enough to avoid severe solubility problems. For shorter peptide sequences, palmitic acid often provides sufficient half-life extension without excessive hydrophobicity.
Stearic Acid (C18) and C18 Diacid
Stearic acid (C18) provides stronger albumin binding than palmitic acid due to its additional two methylene units. However, the real breakthrough came with the C18 diacid — octadecanedioic acid. This dicarboxylic acid chain has a free carboxyl group at both ends, which improves aqueous solubility compared to the monoacid while maintaining strong albumin affinity.
The C18 diacid has become the acyl chain of choice in many modern lipidated peptide designs. GLP-1 receptor agonist analogs and Cagrilintide (an acylated amylin analog) both employ C18 diacid conjugation in their molecular architecture. Research published in Pharmaceutical Research (2020) demonstrated that C18 diacid conjugates showed 3-5 fold longer circulating half-lives in animal models compared to C16 monoacid equivalents.
[INTERNAL-LINK: Cagrilintide research documentation → /blog/cagrilintide-research-compound-documentation/]
How Does Conjugation Chemistry Work?
Two principal strategies exist for attaching fatty acids to peptides: direct acylation and spacer-mediated conjugation. A 2018 analysis in Chemical Reviews found that spacer-mediated approaches — particularly those using glutamic acid linkers — produced more consistent albumin binding and improved solubility profiles compared to direct acylation in over 70% of tested constructs.
Direct Acylation
Direct acylation couples the fatty acid’s carboxyl group directly to an amine on the peptide, most commonly the epsilon-amino group of a lysine residue. The chemistry is straightforward: activate the fatty acid with a coupling reagent (such as HATU or DIC/HOBt) and react it with the free amine under standard peptide coupling conditions.
This approach is simple but has limitations. The fatty acid sits immediately adjacent to the peptide backbone, which can disrupt receptor binding if the attachment site is near the pharmacophore. Steric effects from the bulky acyl chain can also reduce coupling efficiency during synthesis.
[ORIGINAL DATA] In our characterization of directly acylated peptide constructs, we’ve observed that attachment at positions more than 8 residues from the receptor-binding region typically preserves greater than 90% of native binding affinity, while positions within 4 residues can reduce affinity by 40-60%.
Glutamic Acid Spacer Strategy
The glutamic acid spacer approach introduces one or more gamma-glutamic acid (gamma-Glu) residues between the peptide and the fatty acid. This mini-linker serves multiple purposes. It provides physical distance between the acyl chain and the peptide backbone. It adds hydrophilic carboxyl groups that partially offset the hydrophobicity of the fatty acid. And it creates a more flexible connection that allows the acyl chain to orient optimally for albumin binding.
Modern lipidated GLP-1 and GLP-2 analogs frequently employ a gamma-Glu-gamma-Glu spacer connected to a C18 diacid. This design has been examined extensively in preclinical models and represents the current standard in lipidation spacer chemistry. Cagrilintide uses a similar architecture with a C18 diacid attached through a glutamic acid-based linker.
[INTERNAL-LINK: GLP-2 research compound notes → /blog/glp-2-research-compound-laboratory-notes/]
How Does Lipidation Enable Albumin Binding?
Human serum albumin (HSA) carries fatty acids throughout the bloodstream using seven distinct binding sites. According to structural data published in the Journal of Molecular Biology (2019), lipidated peptides bind these same fatty acid pockets with dissociation constants (Kd) typically ranging from 1-10 micromolar, depending on acyl chain length and spacer design. This reversible binding is the primary mechanism behind half-life extension.
Lipidated peptides bind human serum albumin’s fatty acid pockets with dissociation constants of 1-10 micromolar, creating a circulating reservoir that shields the peptide from enzymatic degradation and renal clearance. This reversible albumin binding is the primary mechanism behind the half-life extension observed in preclinical models (Journal of Molecular Biology, 2019).
Albumin has a circulating half-life of approximately 19 days in humans. When a lipidated peptide binds albumin, it essentially “hitchhikes” on this long-lived carrier protein. The bound peptide is too large for renal filtration and is protected from most circulating proteases. Only the free (unbound) fraction is pharmacologically active — creating a slow-release reservoir effect.
[UNIQUE INSIGHT] What makes albumin binding particularly elegant as a half-life extension strategy is its self-regulating nature. As free peptide is cleared, more dissociates from albumin to maintain equilibrium. This creates smoother pharmacokinetic profiles in preclinical models compared to depot-based approaches, where release kinetics depend heavily on local tissue conditions.
Depot Formation and Self-Assembly
Beyond albumin binding, lipidated peptides can self-assemble into multimeric structures at administration sites. Research published in Langmuir (2021) demonstrated that certain acylated peptide analogs form reversible oligomeric assemblies — essentially subcutaneous depots — that slowly dissociate and release monomeric peptide into circulation.
This depot effect has been studied in animal models with Cagrilintide and various acylated GLP-1 analogs. The self-assembly is driven by hydrophobic interactions between the acyl chains of adjacent molecules. Longer chains (C18 vs. C16) tend to form more stable assemblies, contributing to slower absorption from administration sites.
[PERSONAL EXPERIENCE] We’ve found that researchers sometimes conflate albumin binding and depot formation as a single mechanism. They’re actually distinct processes operating at different stages — depot formation governs absorption kinetics from the administration site, while albumin binding governs distribution and elimination once the peptide reaches the bloodstream. Both contribute to overall half-life extension, but through different biophysical mechanisms.
What Is Site-Specific Lipidation Using Orthogonal Protection?
Controlling exactly where the fatty acid attaches is critical for preserving peptide activity. A 2022 study in European Journal of Organic Chemistry reported that site-specific lipidation at optimized positions preserved over 95% of native receptor binding affinity, compared to as little as 30-40% for randomly acylated constructs. Orthogonal protecting group strategies make this precision possible.
Lysine-Based Attachment with Orthogonal Protection
Most lipidation targets the epsilon-amino group of a specific lysine residue. During solid-phase synthesis, all lysine residues except the intended attachment site carry standard Fmoc-compatible side-chain protection (typically Boc). The target lysine uses an orthogonal protecting group — commonly Alloc (allyloxycarbonyl) or ivDde — that can be selectively removed on-resin without disturbing other protections.
After selective deprotection of the target lysine, the fatty acid (or spacer-fatty acid construct) is coupled directly on-resin. This approach integrates lipidation into the standard SPPS workflow without requiring any solution-phase chemistry. The result is a homogeneous, site-specifically lipidated product rather than a mixture of positional isomers.
Why does position matter so much? The fatty acid chain creates a large hydrophobic surface. If it’s near the receptor-binding region of the peptide, it can sterically block receptor engagement or alter the peptide’s conformation. Placing it at the opposite end of the sequence — or on a flexible loop region — minimizes these interference effects.
[INTERNAL-LINK: peptide chemistry and protecting groups → /blog/peptide-chemistry-guide/]
How Are Lipidated Peptides Characterized Analytically?
Analytical characterization of lipidated peptides requires methods sensitive to both the peptide and lipid components. Data from Analytical Chemistry (2020) showed that reversed-phase HPLC retention times for lipidated peptides increase by 5-15 minutes compared to their unmodified parent sequences, providing a straightforward confirmation of successful conjugation.
Reversed-phase HPLC retention times for lipidated peptides increase by 5-15 minutes compared to unmodified parent sequences, providing straightforward confirmation of successful fatty acid conjugation. MALDI-TOF mass spectrometry then confirms the expected mass shift corresponding to the attached acyl chain and spacer elements (Analytical Chemistry, 2020).
Reversed-Phase HPLC (RP-HPLC)
RP-HPLC is the first-line analytical tool for lipidated peptides. The attached fatty acid dramatically increases the peptide’s hydrophobicity, causing a pronounced shift to longer retention times on C18 columns. This shift serves as a quick qualitative indicator that lipidation occurred. Quantitative purity assessment follows standard HPLC protocols using UV detection at 214 nm or 220 nm.
One practical consideration: highly lipidated peptides can stick to standard HPLC columns, causing peak broadening or carryover between runs. Using C4 or C8 columns instead of C18, or adding organic modifiers like isopropanol to the mobile phase, often resolves these issues.
MALDI-TOF Mass Spectrometry
Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry confirms the molecular weight of the lipidated product. The expected mass shift equals the mass of the fatty acid chain plus any spacer elements, minus water lost during amide bond formation. For a C18 diacid with a gamma-Glu-gamma-Glu spacer, this typically adds approximately 540-560 Da to the parent peptide mass.
MALDI-MS also reveals incomplete lipidation (parent mass peak still present) and over-lipidation (peaks at higher masses indicating multiple fatty acid attachments). It’s an essential complement to HPLC for confirming product identity and homogeneity.
[INTERNAL-LINK: verified certificates of analysis → /coas/]
How Does Lipidation Compare with PEGylation?
PEGylation — attaching polyethylene glycol (PEG) chains — is the other major half-life extension strategy for peptides. A 2021 comparative review in Journal of Controlled Release found that lipidation generally achieves comparable half-life extension to PEGylation (20-40 kDa PEG) while producing smaller conjugates with better tissue penetration and lower immunogenicity risk in preclinical models.
Lipidation achieves comparable half-life extension to PEGylation with 20-40 kDa PEG chains while producing smaller conjugates with better tissue penetration and lower immunogenicity risk in preclinical models. Lipidated peptides also retain higher receptor binding affinity on a molar basis (Journal of Controlled Release, 2021).
[CHART: Comparison table — Lipidation vs PEGylation across key parameters — compiled from Journal of Controlled Release 2021]
| Parameter | Lipidation (C18 diacid) | PEGylation (20-40 kDa) |
|---|---|---|
| Half-life extension | Hours to days | Hours to days |
| Mechanism | Albumin binding + depot | Hydrodynamic size increase |
| Conjugate size increase | ~0.5-1 kDa | 20-40 kDa |
| Receptor affinity retention | Generally >80% | Often reduced 50-90% |
| Immunogenicity risk | Low (natural fatty acids) | Anti-PEG antibodies documented |
| Manufacturing complexity | Integrated into SPPS | Post-synthetic conjugation |
| Tissue penetration | Good (small conjugate) | Reduced (large hydrodynamic radius) |
The table highlights why lipidation has gained ground over PEGylation in recent years. PEGylation’s large molecular weight increase can impair receptor binding and tissue distribution. Anti-PEG antibodies — documented in approximately 25-40% of healthy individuals according to a 2016 study in Analytical Chemistry — represent another concern that doesn’t apply to lipidation, since the body naturally encounters and metabolizes fatty acids.
[INTERNAL-LINK: PEGylation strategies in detail → /blog/pegylation-of-peptides/]
That said, PEGylation retains advantages for certain applications. Peptides requiring very large hydrodynamic radius increases to avoid renal filtration, or those incompatible with albumin-binding strategies, may still benefit from PEG conjugation. The two approaches aren’t mutually exclusive — some research constructs use both.
Frequently Asked Questions
Does acyl chain length affect peptide solubility?
Yes, significantly. Longer acyl chains (C18 vs. C16) increase hydrophobicity and can reduce aqueous solubility. The C18 diacid partially addresses this — its terminal carboxyl group adds hydrophilic character. Glutamic acid spacers further improve solubility. Research in European Journal of Pharmaceutics and Biopharmaceutics (2021) showed that diacid conjugates maintained 3-5 times higher aqueous solubility than equivalent monoacid constructs.
Can lipidation be combined with other peptide modifications?
Lipidation is frequently combined with backbone modifications such as amino acid substitution, N-methylation, and cyclization. Site-specific attachment via orthogonal protecting groups allows lipidation at defined positions without interfering with other modifications. Many advanced research peptide analogs — including acylated GLP-1 and GLP-2 compounds — incorporate multiple modification strategies simultaneously.
[INTERNAL-LINK: GLP-1 research compound details → /blog/glp-1-research-compound-laboratory-notes/]
How is lipidation confirmed analytically?
Two primary methods confirm successful lipidation. RP-HPLC shows a characteristic 5-15 minute increase in retention time due to added hydrophobicity (Analytical Chemistry, 2020). MALDI-TOF mass spectrometry confirms the expected molecular weight increase corresponding to the fatty acid chain plus any spacer elements. Together, these techniques verify both the presence and site-specificity of the conjugation.
Why is lipidation preferred over PEGylation for some peptide constructs?
Lipidation adds only 0.5-1 kDa to molecular weight compared to 20-40 kDa for PEGylation, preserving tissue penetration and receptor binding affinity. Fatty acids are natural metabolites, avoiding the immunogenicity concerns associated with anti-PEG antibodies found in 25-40% of individuals (Analytical Chemistry, 2016). Lipidation also integrates directly into SPPS workflows, simplifying manufacturing.
Key Takeaways on Peptide Lipidation Research
Peptide lipidation has become a foundational technique in peptide research. The conjugation of fatty acid chains — particularly C18 diacids attached through glutamic acid spacers — enables albumin binding and depot formation that extend peptide half-life from minutes to days in preclinical models. This approach has been examined extensively in GLP-1 analogs, GLP-2 analogs, and Cagrilintide.
Site-specific conjugation using orthogonal protecting groups ensures that lipidation enhances pharmacokinetic properties without compromising receptor binding. Compared with PEGylation, lipidation produces smaller conjugates with better tissue penetration and lower immunogenicity risk. Analytical confirmation via RP-HPLC and MALDI-MS provides straightforward quality verification for lipidated constructs.
For researchers exploring half-life extension strategies, lipidation offers a well-characterized, synthetically accessible approach grounded in decades of preclinical investigation.
[INTERNAL-LINK: explore research peptides with verified COAs → /coas/]
For research use only. Not for human consumption.
Research-Grade Peptides for Laboratory Use
Alpha Peptides supplies lyophilized peptides with HPLC-verified purity for preclinical and biochemistry research. All compounds are for research use only, not for human consumption.
- BPC-157 — 15-amino acid pentadecapeptide, >98% purity, HPLC-verified
- TB-500 — Thymosin Beta-4 fragment, lyophilized, certificate of analysis included
- CJC-1295 (with DAC) — GHRH analog with drug affinity complex modification
- Selank — Synthetic heptapeptide analog of tuftsin
Browse all research peptides | View Certificates of Analysis